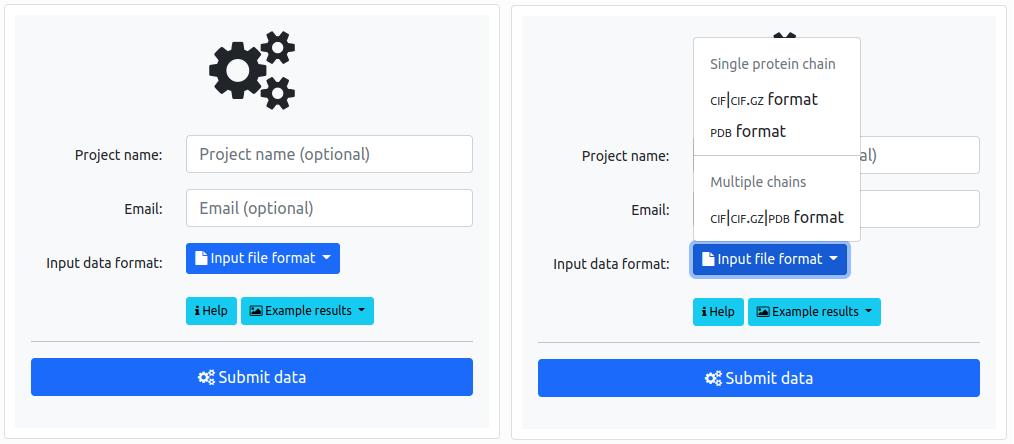
A user can upload and analyze two types of data: either a single structure, or a set of structures – possibly models of the same protein – whose topology one wants to compare. The "Input file format" button provides these two options. In this section we explain how to analyze a single structure.
The user can choose several options which determine what methods will be used to describe the entanglement in the structure, and also determine the accuracy of the calculations. The calculation time on the server is directly related to the selected parameters (and to the length and complexity of the structure). For a single structure, it varies from a few seconds to several minutes. Therefore, choosing the correct options is important.
The key options to choose from are visible after expanding the "Calculate" list:
Lasso and matrix (default option) - means that the server will first check to see if the structure has loops. If it does, the lasso GLN map is computed.
Lasso calculation only - means that the server will find the disulphide bridges and determine the type of the loop. It will also generate a surface spanned on that loop.
To detect a loop type, a chain must have a closing bridge (for more details see section Lasso detection). A user can choose from two options:
Automatic
Specific aminoacids
Choose your own loop closing bridges
The first method involves analyzing the model for the presence of cysteine, amide, ester, thioester bonds. In the second mode, user provides two aminoacids that form a bridge (by their three letter code). By choosing the third option, user can manually define indexes of residue for loop closing.
Homologs can be searched in the Database on the selected by the user identity level, between 70%, 80%, or 90%. This option has a significant impact on the calculation time. If you do not need information about homologs, you can disable this search.
By clicking Show advanced button, user can access additional settings. In the bridge length section the minimum and maximum distance between bridge atoms can be specified. When using the Automatic mode the distance is measured between atoms specific for each bond. In the other two modes it is measured between alpha carbons of the specifed aminoacids. It is possible to define the number of smoothing iterations (must be greater than 0 to generate barycentric view), and after unchecking the Stable lasso checkbox, the minimal distances regarding crossings can be set. By default, for the similarity search, only non-L0 models are considered, and an AlphaFold/Proteome Foldseek database is used. After checking Search for identity in all AlphaLasso and AlphaFold models, the search will be conducted through the entire AlphaLasso database and AlphaFold/UniProt50 Foldseek database.
A structure uploaded and analyzed by a user is stored for 14 days (With the exception of structures uploaded via the recompute tool, which will update the corresponding record in the Database).
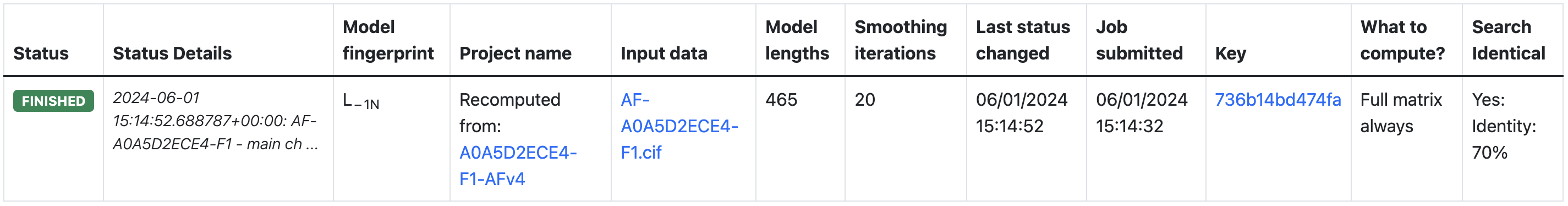
At the top of the output, there is a table that contains the status details of the job, information about main methods and parameters used for that job, and the main results. The page will automatically refresh to track the progress of the job.
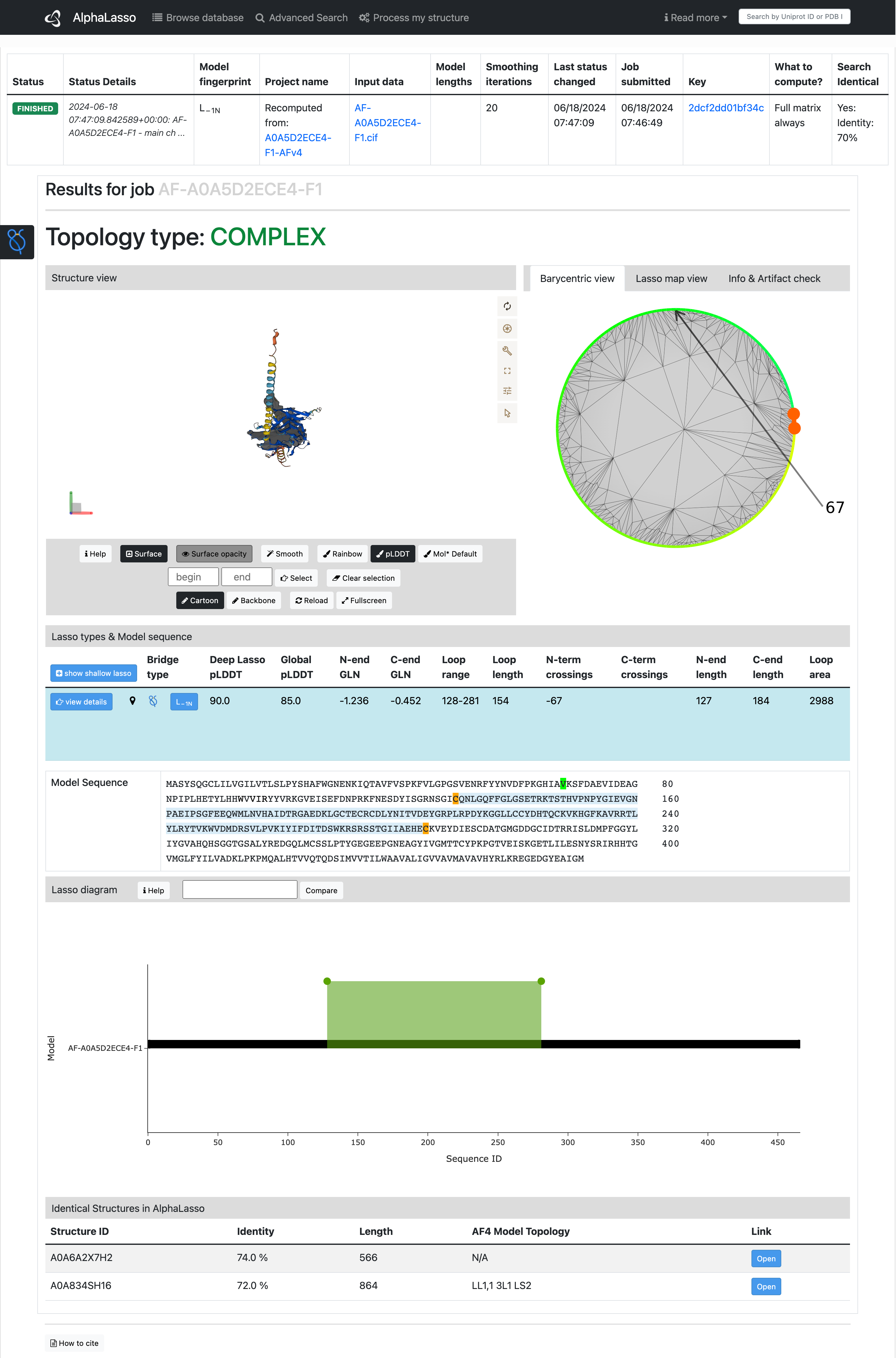
When all the results are ready, there is information on whether the structure's topology is complex. If the structure has loops, there is a view similar to the one for each record in the database. This view is divided into four main parts:
View of the structure
Barycentric view – the view is not visible if the smoothing iterations value was set to 0.
Lasso map – the map is not visible if the matrix option was not chosen.
Information and Artifact check tables The information table consists of basic information on the topology of the structure (Loop fingerprint) and about the quality of whole structure – the average pLDDT for whole structure (Global pLDDT). Artifact check table gives results of some automatic tests which can help the user to recognize if the topology may be an artifact. The table may not be displayed if the input file does not contain the information about pLDDT. The value of pLDDT measures the confidence of the predicted position of particular amino acid.
All CIF or PDB files with structures predicted by AlphaFold do contain pLDDT values.
C-alpha clashes controls if there are parts of chain which are located at a distance shorter than 1.5A from each other.
pLDDT model is the average pLDDT value of the whole model.
pLDDT loop is the average pLDDT value of the loop region (part of the chain where the loop is located); values below 50 are a strong indication that the topology is an artifact; values below 70 suggest thet the result should be interpreted with extreme caution.
In some cases there may be both deep and shallow lassos depending on which crossings one takes into account. The appropriate pLDDT value is displayed depending on whether or not the "Show Shallow Lasso" is pressed.
pLDDT N-end/C-end loop tails are the average pLDDT values of tail residues from the loop up to the last crossing.
If there is any information in red in this table, the user should be leery about the results.
Lasso types and model sequence
List of similar proteins in the database with their topology
The similar proteins are searched for using both MMseqs2 and Foldseek. The method used is listed in the table.
If the structure has no loops then the lasso map and the structure are not displayed.
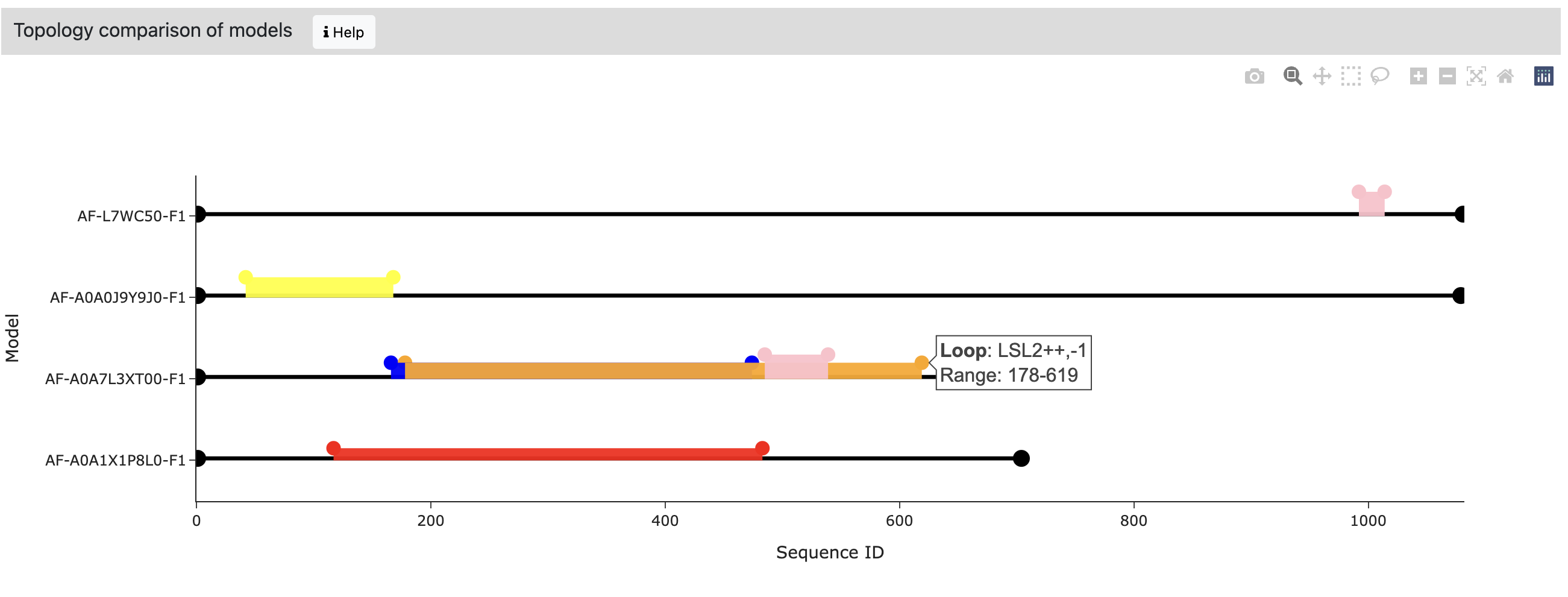
Up to 10 files in CIF or PDB format can be uploaded at once. These can be different models of the same structure (as in this example) or any other set of structures. In this case, at the top of the output page, we can select the model / structure for which we want to see the results.
And at the bottom of the view, we additionally see a comparison of the location and types of loops in each structure. The color of the bar corresponds to the type of loop. When the user hovers the mouse over the dots ending the colored bars, information about the loop is shown.
The AlphaLasso user can use recompute tool to redirect structures currently available in the database to the server to obtain additional information. User can control the parameters of the calculation similar to the direct structure upload described above. The additional features visible after structure recompute will be shown on the model page only if the default values were used.